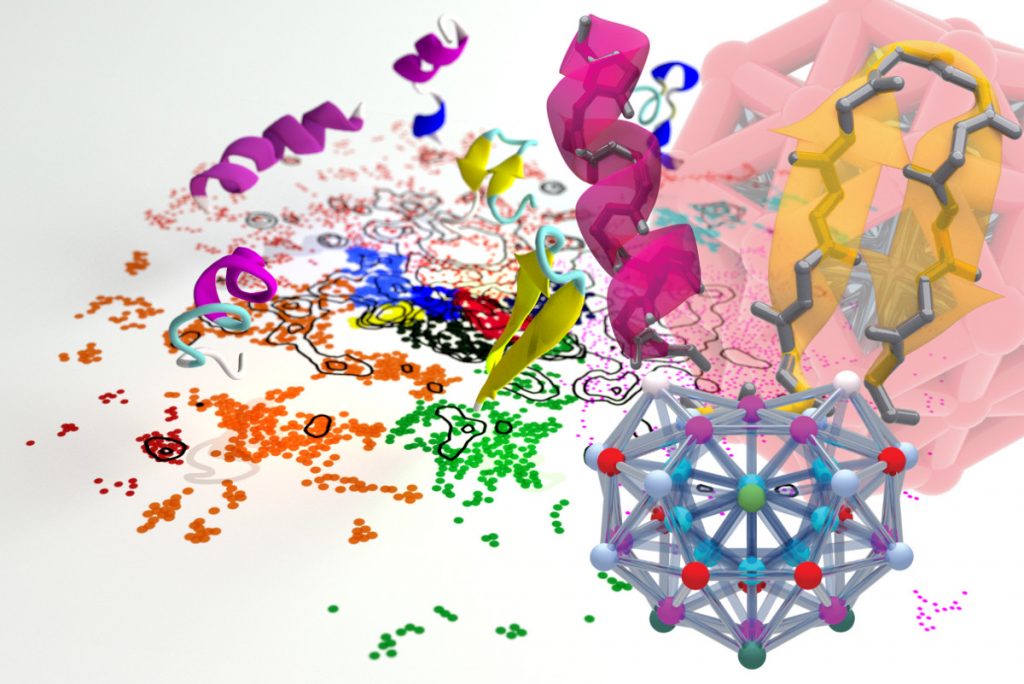
Machine-learning Analysis of Molecular Patterns
What is a chemical bond? How do we recognize that an atom has a preferred coordination environment? Atomistic simulations obtain an accurate description of the arrangement of atoms as dictated by their interactions. Often, the real problem is to determine which patterns appear more often in a simulation, so that they can be used to rationalize the behavior of a material or a molecolar compound.
A good example is that of the hydrogen bond. Chemists have discussed for a century about how to rationalize this ubiquitous entity composed by two electro-negative atoms and a hydrogen, that spans a broad range of binding energies going from a fraction of a kcal/mol, to several tens. Rather than defining more or less arbitrary ranges of structural parameters associated with a bond, or to use complicated energy decomposition methods, we tackle this problem by analyzing automatically the configurations obtained in a simulation. We recognize recurring patterns by locating local maxima in the probability distribution of the relative positions of the donor, acceptor and hydrogen atoms, and obtain a Gaussian Mixture Model representation that naturally engenders a probabilistic, unbiased definition of what can be deemed to represent a (hydrogen) bond.
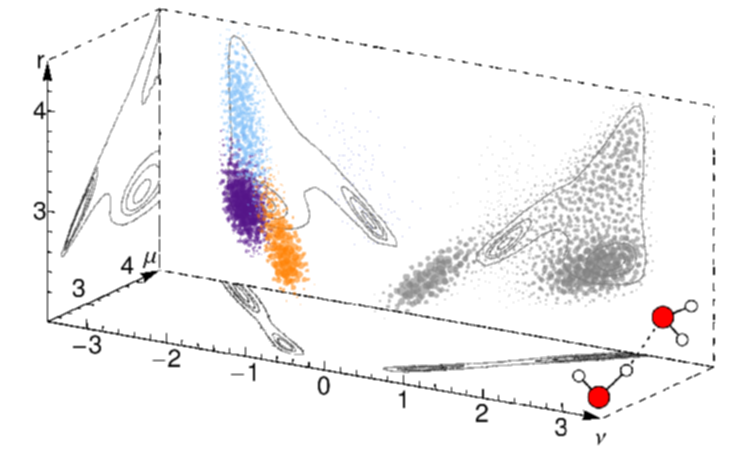
This Probabilistic Analysis of Molecular Motifs (PAMM) technique is completely general, and we are currently exploring its use in recognizing coordination patterns in solids, and the prevalence of local structural motifs in polymers.
Structural complexity in (bio)-polymers
When performing simulations containing thousands or millions of atoms, describing accurately the motion of the different components of the system is not the only problem one must face. The sheer amount of data produced by these simulations makes it very hard to understand what goes on, and it is often useful to use the computer also to perform a post-processing analysis.
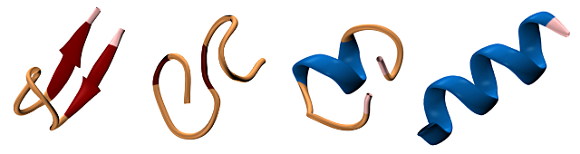
These problems are particularly challenging when one is simulating structurally-complex materials, such as glasses, polymers or proteins: while on a macroscopic scale we can often interpret these compounds as existing in a few, relatively well-defined thermodynamic states (amorphous/crystalline, folded/denatured, …) on a microscopic scale there are billions of possible configurations, differing by the coordinates of some of the atoms.

A possible approach to tackle these problems uses non-linear dimensionality reduction techniques (see e.g. sketchmap.org) to find a 2D representation of the problem, a “map” that can be used to make sense of the results of simulations and relate them to phenomenological observations.
These machine learning techniques are not only useful to interpret the result of simulations, but can also be used to perform accelerated sampling simulations, that try to bridge the enormous gap that exists between the time scale accessible to simulations and the time scale that is relevant to experiments.