High-Quality Data Enabling Universality of Band Gap Descriptor and Discovery of Photovoltaic Perovskites
Haiyuan Wang, Runhai Ouyang, Wei Chen
and Alfredo Pasquarello
J. Am. Chem. Soc. 146, 17636-17645 (2024)

A dataset of high-quality band gaps for perovskite materials is first generated through an advanced electronic structure method and then used to develop a simple but accurate band-gap model through artificial intelligence. This enables a search across large databases leading to the identification of promising halide perovskites for photovoltaic applications.
Published on 3 May 2024
Dynamics of the charge transfer to solvent process in aqueous iodide
Jinggang Lan, Majed Chergui, and Alfredo Pasquarello
Nat. Commun. 15, 2544 (2024)

Charge-transfer-to-solvent states in aqueous halides are ideal systems for studying the electron-transfer dynamics to the solvent involving a complex interplay between electronic excitation and solvent polarization. However, despite extensive experimental investigations, a full picture of the charge-transfer-to-solvent dynamics has remained elusive. This study combines ab initio molecular dynamics and machine learning to visualize the evolution from the electron excitation until its full solvation. A sequence of intermediate states is identified and supported by the excellent agreement with energy spectra from time-resolved photoelectron spectroscopy measurements.
Published on
Absolute energy levels of liquid water from many-body perturbation theory with effective vertex corrections
Alexey Tal, Thomas Bischoff, and Alfredo Pasquarello
Proc. Natl. Acad. Sci. U.S.A. 121, e2311472121 (2024)

The knowledge of the electronic structure of liquid water is essential for fundamental science and technology. However, state-of-the-art electronic-structure schemes have so far been unable to match experimental energy levels, by which severe ambiguities persist for the ionization potential and the electron affinity of liquid water. Here, it is shown that the consideration of a vertex function within many-body perturbation theory succeeds in producing photoemission and absorption spectra in excellent agreement with experiment on the absolute scale, overcoming this long standing issue.
Published on 1 March 2024
Many-body screening effects in liquid water
Igor Reshetnyak, Arnaud Lorin,
and Alfredo Pasquarello
Nat. Commun. 14, 2705 (2023)

The screening arising from many-body excitations is a crucial quantity for describing absorption and inelastic X-ray scattering (IXS) of materials. Similarly, the electron screening plays a critical role in state-of-the-art approaches for determining the fundamental band gap. However, ab initio studies of the screening in liquid water have remained limited. Here, we use a combined analysis based on the Bethe-Salpeter equation and time-dependent density functional theory. We first show that absorption spectra at near-edge energies are insufficient to assess the accuracy by which the screening is described. Next, when the energy range under scrutiny is extended, we instead find that the IXS spectra are highly sensitive and allow for the selection of the optimal theoretical scheme. This leads to good agreement with experiment over a large range of transferred energies and momenta, and enables establishing the elusive fundamental band gap of liquid water at 9.3 eV.
Published on 11 May 2023
Many-body self-interaction and polarons
Stefano Falletta and Alfredo Pasquarello
Phys. Rev. Lett. 129, 126401 (2022)
Phys. Rev. B 106, 125119 (2022)
Semilocal density functional theory generally fails in stabilizing polarons because of its spurious inclusion of the electron self-interaction. This work addresses the concepts of many-body and one-body self-interaction through the development of a unified theoretical framework. This is instrumental to establish a quantitative connection between these two forms of self-interaction through the role of the electron screening, whereby the many-body self-interaction is identified as a superior concept. Next, the polaron self-interaction is addressed through the nonempirical construction of piecewise-linear functionals at the semilocal level of theory. Polaron properties, such as charge densities, lattice distortions, and formation energies are found to be robust upon variation of the functional adopted and in good agreement with reference hybrid functional results. Hence, this work not only advances the conceptual understanding of the self-interaction problem in density functional theory, but also achieves an efficient computational description of polaronic states.
Published on 14 September 2022
Temperature dependent properties of the aqueous electron
Jinggang Lan, Vladimir V. Rybkin,
and Alfredo Pasquarello
Angew. Chem. Int. Ed. 61, e202209398 (2022)
The temperature-dependent properties of the aqueous electron have been extensively studied using mixed quantum-classical simulations in a wide range of thermodynamic conditions based on one-electron pseudopotentials. While the cavity model appears to explain most of the physical properties of the aqueous electron, only a non-cavity model has so far been successful in accounting for the temperature dependence of the absorption spectrum. Here, we present an accurate and efficient description of the aqueous electron under various thermodynamic conditions by combining hybrid functional-based molecular dynamics, machine learning techniques, and multiple time-step methods. Our advanced simulations accurately describe the temperature dependence of the absorption maximum in the presence of cavity formation. Specifically, our work reveals that the red shift of the absorption maximum results from an increasing gyration radius with temperature, rather than from global density variations as previously suggested.
Published on 18 July 2022
Evaluation of photocatalysts for water splitting through combined analysis of surface coverage and energy-level alignment
Zhendong Guo, Francesco Ambrosio
and Alfredo Pasquarello
ACS Catal. 10, 13186−13195 (2020)
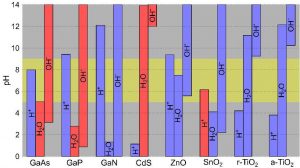
To examine whether suitable conditions occur for the water splitting reaction at their interfaces with liquid water, we determine the pH-dependent surface coverage for a series of semiconductors. For this we calculate acidity constants at surface sites through ab initio molecular dynamics simulations and a grand-canonical formulation of adsorbates. The resulting pH values at the point of zero charge show excellent agreement with experiment and thereby support the validity of our approach. By combining information concerning the surface coverage with the alignment of the band edges with respect to the relevant redox levels, we scrutinize the potential of the considered semiconductors as photocatalysts and identify the corresponding optimal pH ranges for hydrogen and oxygen evolution. With the surface coverage, our computational analysis brings a new descriptor that is at present beyond experimental reach.
Published on 30 October 2020
Picture of the wet electron: a localized transient state in liquid water
Chem. Sci. 10, 7442-7448 (2019)

A transient state of the excess electron in liquid water preceding the development of the solvation shell, the so-called wet electron, has been invoked to explain spectroscopic observations, but its binding energy and atomic structure have remained highly elusive. Here, we carry out hybrid functional molecular dynamics to unveil the ultrafast solvation mechanism leading to the hydrated electron. In the pre-hydrated regime, the electron is found to repeatedly switch between a quasi-free electron state in the conduction band and a localized state with a binding energy of 0.26 eV, which we assign to the wet electron. This transient state self-traps in a region of the liquid which extends up to ∼4.5 Å and involves a severe disruption of the hydrogen-bond network. Our picture provides an unprecedented view on the nature of the wet electron, which is instrumental to understanding the properties of this fundamental species in liquid water.
Published on 26 June 2019