The project TARGET aims at developing and applying a novel strategy for targeting so-called ‘undruggable’ proteins by tapping into a new chemical space, generated by ‘merging’ biological and chemical compound libraries. The new methods should deliver synthetic ligands to notoriously difficult-to-target proteins as for example challenging intracellular protein-protein interactions. Such ligands would offer ideal starting points for developing new therapeutics to address important unmet medical needs.
The problem we address
Many important drug targets are not amenable to small molecule ligands. Genome sequencing combined with powerful new technologies, ranging from proteomic methods to genome editing tools for generating transgenic animals, are constantly providing new potential drug targets. However, many of these proteins are not amenable to classical small molecule drugs, the mainstay of drug development. Small molecules are particularly important for intracellular proteins due to their cell permeability, as well as for extracellular targets if the drug needs to be administered orally. However, small molecules rely on deep clefts or pockets in the protein to bind with sufficient affinity, yet many key proteins or interactions lack this necessary pocket. Prominent examples of challenging proteins that have been difficult to target include TNFalpha, KRASG12D, PCSK9, MYC, NF-κB, AR-V7, VEGF, IL-23R, to name a few.
Macrocyclic molecules may offer a promising solution to the currently ‘undruggable’ disease targets. Due to their cyclic configuration, macrocyclic compounds have rather limited conformational flexibility, which reduces the entropic penalty of binding to targets, resulting in good binding affinities with a relatively small molecular footprint. If they are small enough (well below 1000 Da) and have a limited polar surface area, they typically can cross membranes, allowing access to intracellular targets and oral delivery. Unfortunately, it is difficult to develop de novo macrocyclic molecules that bind to new disease targets, mainly due to the lack of sufficiently large compound libraries and methods to identify binders in such libraries (‘needle in a haystack’ problem), as described in the following paragraph.
The development of ligands based on macrocyclic compounds is currently difficult due to the lack of sufficiently large libraries of this type of structure. Biological display methods such as phage display can in principle be used to screen reasonably large libraries of small cyclic peptides (e.g. 6-mers: 20e6 = 64,000,000 cyclic peptides). However, these libraries contain only the 20 canonical amino acids as building blocks and therefore have a chemical diversity that is too low to find binders for challenging targets. The situation is slightly better with mRNA display libraries, which allow the insertion of non-natural amino acids. However, the lack of codons and technical challenges limit the number of different amino acids that can be inserted combinatorially. To achieve high binding affinities with small cyclic structures, each chemical group of the cyclic peptide must contribute substantially to the binding affinity (ligand efficiency), but this can only be achieved if the number of different building blocks sampled is much larger than that of existing phage or mRNA display libraries. Chemical methods allow generating structurally and chemically much more diverse molecules but there are currently no efficient methods to generate and screen large libraries of chemically synthesized macrocyclic compounds.
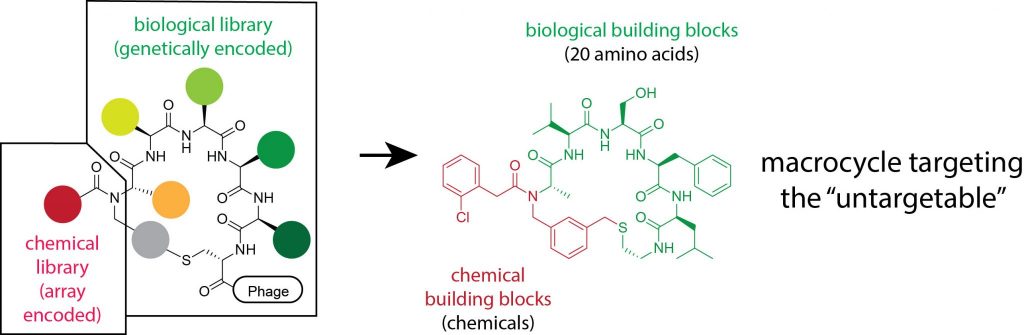
Solution that we propose and strategy that we follow
The core idea of the TARGET project is to merge biological and chemical compound libraries, the first library type being generated by ribosomal translation (e.g. phage display peptide libraries) and the second one by chemical synthesis. The two types of libraries have their strengths and weaknesses and were so far used mostly independently from each other in drug development. With biological libraries, it is possible to generate and screen more than a billion random peptides but the chemical diversity of these libraries is small (e.g. in phage display libraries, the building blocks are mostly limited to the 20 natural amino acids). In contrast, chemical libraries are structurally highly diverse and can be generated using a nearly endless number of building blocks, but the number of molecules that can be handled and screened is limited. In the TARGET project, biological compound libraries (phage-displayed peptides) are chemically diversified with large numbers of chemical building blocks of structurally and chemically highly diverse fragments, to generate and screen a previously unseen large and structurally highly diverse chemical space.
Importance for the society
With the planned research, we address a major gap in drug development being the modulation of challenging intracellular disease targets that have proven difficult or impossible to address by classical small molecule drugs. Macrocyclic compounds hold promise as a solution to this problem, as demonstrated by naturally derived cyclic peptides that can enter cells and exert biological functions. We propose developing and applying a strategy to generate cyclic peptide-based ligands of some of the most important but also most challenging targets. If successful, the membrane permeable ligands/inhibitors have a high chance to be translated into therapeutics. Given the implication of the chosen targets in a wide range of abundant cancer types, a large number of patients could potentially benefit of the results. A proof-of-concept demonstration for the strategy would likely also trigger the broad application of the technologies for many more targets, both in academia and industry, and ligands/inhibitors may be developed to many proteins and diseases with an enormous potential impact on patients and society.