S. Jawahery, N. Rampal, S. M. Moosavi, M. Witman, and B. Smit, Ab Initio Flexible Force Field for Metal-Organic Frameworks Using Dummy Model Coordination Bonds J. Chem. Theory Comput. (2019) Doi:10.1021/acs.jctc.9b00135
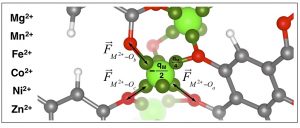
Abstract: We present force fields developed from periodic density functional theory (DFT) calculations that can be used in classical molecular simulations to model M-MOF-74 (M = Co, Fe, Mg, Mn, Ni, Zn) and its extended linker analogs. Our force fields are based on cationic dummy models (CDMs). These dummy models simplify the methodology required to tune the parameters and improve the accuracy of the force fields. We have used our force fields to compare mechanical properties across the M-MOF-74 series, and determine that increasing the size of the linker decreases the framework rigidity. In addition, we have applied our force fields to an extended linker analog of Mg-MOF-74 and characterized the free energy of previously-reported deformation pattern, in which the one-dimensional hexagonal channels of the framework become irregular. The free energy profiles confirm that the deformation is adsorbate-induced and impossible to access solely by a pressure stimulus. Based on our results, we conclude that the force fields presented here and others that may be developed using our methodology are transferable across metal-organic framework series that share a metal center topology. Finally, we believe that these force fields have the potential to be adapted for the study of complex problems in MOF chemistry, including defects and crystal growth, that have thus far been beyond the scope of classical molecular simulations.