H. Gustafsson, M. Kozdra, B. Smit, S. Barthel, and A. Mace, Predicting Ion Diffusion from the Shape of Potential Energy Landscapes J. Chem. Theory Comput. (2023) DOI: 10.1021/acs.jctc.3c01005
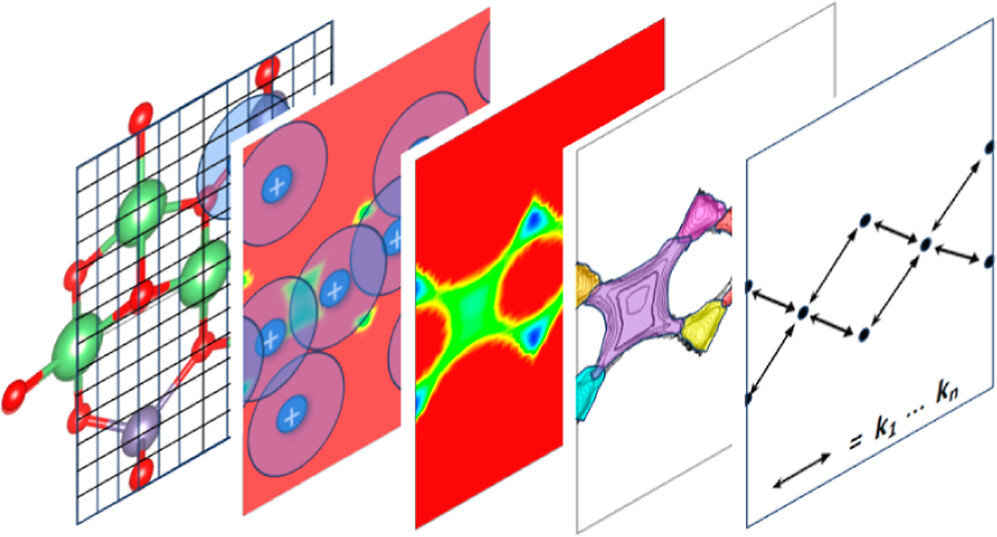
Abstract: We present an efficient method to compute diffusion coefficients of multiparticle systems with strong interactions directly from the geometry and topology of the potential energy field of the migrating particles. The approach is tested on Li-ion diffusion in crystalline inorganic solids, predicting Li-ion diffusion coefficients within 1 order of magnitude of molecular dynamics simulations at the same level of theory while being several orders of magnitude faster. The speed and transferability of our workflow make it well-suited for extensive and efficient screening studies of crystalline solid-state ion conductor candidates and promise to serve as a platform for diffusion prediction even up to the density functional level of theory.