Projects
Our lab is interested in quantitative, systems biology, and more generally the link between physics and biology. We work on various problems including circadian rhythms, transcriptional bursting, developmental patterning, gene regulation, and single cell imaging. To study these systems we combine theoretical, computational and experimental methods.
Current projects:
- Circadian gene regulatory networks in mammals
- Chronobiology of the liver
- Circadian oscillators in single cells
- Interactions of circadian oscilators and cell cycle
- Transcriptional bursting and noise in mammalian gene transcription
Research highlights (cont’)

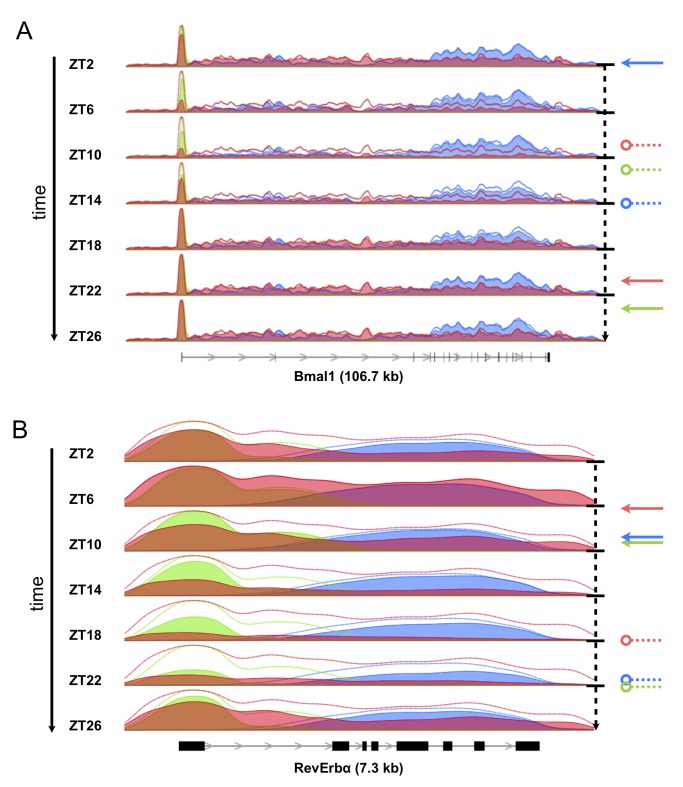
April 2017: Transcriptional regulatory logic of the diurnal cycle in the mouse liver. Many organisms exhibit temporal rhythms in gene expression that propel diurnal cycles in physiology. In the liver of mammals, these rhythms are controlled by transcription–translation feedback loops of the core circadian clock and by feeding–fasting cycles. To better understand the regulatory interplay between the circadian clock and feeding rhythms, we mapped DNase I hypersensitive sites (DHSs) in the mouse liver during a diurnal cycle. The intensity of DNase I cleavages cycled at a substantial fraction of all DHSs, suggesting that DHSs harbor regulatory elements that control rhythmic transcription. Using chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq), we found that hypersensitivity cycled in phase with RNA polymerase II (Pol II) loading and H3K27ac histone marks. We then combined the DHSs with temporal Pol II profiles in wild-type (WT) and Bmal1-/- livers to computationally identify transcription factors through which the core clock and feeding–fasting cycles control diurnal rhythms in transcription. While a similar number of mRNAs accumulated rhythmically in Bmal1-/- compared to WT livers, the amplitudes in Bmal1-/- were generally lower. The residual rhythms in Bmal1-/- reflected transcriptional regulators mediating feeding–fasting responses as well as responses to rhythmic systemic signals. Finally, the analysis of DNase I cuts at nucleotide resolution showed dynamically changing footprints consistent with dynamic binding of CLOCK:BMAL1 complexes. Structural modeling suggested that these footprints are driven by a transient heterotetramer binding configuration at peak activity. Together, our temporal DNase I mappings allowed us to decipher the global regulation of diurnal transcription rhythms in the mouse liver.
• Publisher site • Pubmed • Web of Science • Infoscience
 October 2016: Nuclear Proteomics Uncovers Diurnal Regulatory Landscapes in Mouse Liver. Diurnal oscillations of gene expression controlled by the circadian clock and its connected feeding rhythm enable organisms to coordinate their physiologies with daily environmental cycles. While available techniques yielded crucial insights into regulation at the transcriptional level, much less is known about temporally controlled functions within the nucleus and their regulation at the protein level. Here, we quantified the temporal nuclear accumulation of proteins and phosphoproteins from mouse liver by SILAC proteomics. We identified around 5,000 nuclear proteins, over 500 of which showed a diurnal accumulation. Parallel analysis of the nuclear phosphoproteome enabled the inference of the temporal activity of kinases accounting for rhythmic phosphorylation. Many identified rhythmic proteins were parts of nuclear complexes involved in transcriptional regulation, ribosome biogenesis, DNA repair, and the cell cycle and its potentially associated diurnal rhythm of hepatocyte polyploidy. Taken together, these findings provide unprecedented insights into the diurnal regulatory landscape of the mouse liver nucleus.
October 2016: Nuclear Proteomics Uncovers Diurnal Regulatory Landscapes in Mouse Liver. Diurnal oscillations of gene expression controlled by the circadian clock and its connected feeding rhythm enable organisms to coordinate their physiologies with daily environmental cycles. While available techniques yielded crucial insights into regulation at the transcriptional level, much less is known about temporally controlled functions within the nucleus and their regulation at the protein level. Here, we quantified the temporal nuclear accumulation of proteins and phosphoproteins from mouse liver by SILAC proteomics. We identified around 5,000 nuclear proteins, over 500 of which showed a diurnal accumulation. Parallel analysis of the nuclear phosphoproteome enabled the inference of the temporal activity of kinases accounting for rhythmic phosphorylation. Many identified rhythmic proteins were parts of nuclear complexes involved in transcriptional regulation, ribosome biogenesis, DNA repair, and the cell cycle and its potentially associated diurnal rhythm of hepatocyte polyploidy. Taken together, these findings provide unprecedented insights into the diurnal regulatory landscape of the mouse liver nucleus.
Publisher site • Pubmed • Web of Science • Infoscience
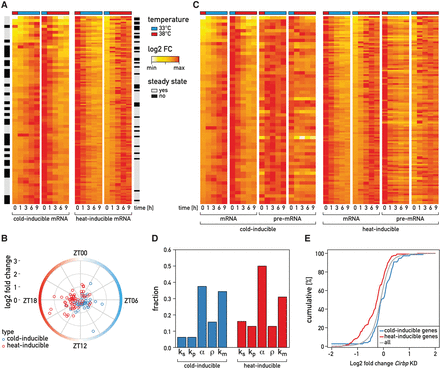 September 2016: Temperature regulates splicing efficiency of the cold-inducible RNA-binding protein gene Cirbp. In mammals, body temperature fluctuates diurnally around a mean value of 36°C-37°C. Despite the small differences between minimal and maximal values, body temperature rhythms can drive robust cycles in gene expression in cultured cells and, likely, animals. Here we studied the mechanisms responsible for the temperature-dependent expression of cold-inducible RNA-binding protein (CIRBP). In NIH3T3 fibroblasts exposed to simulated mouse body temperature cycles, Cirbp mRNA oscillates about threefold in abundance, as it does in mouse livers. This daily mRNA accumulation cycle is directly controlled by temperature oscillations and does not depend on the cells’ circadian clocks. Here we show that the temperature-dependent accumulation of Cirbp mRNA is controlled primarily by the regulation of splicing efficiency, defined as the fraction of Cirbp pre-mRNA processed into mature mRNA. As revealed by genome-wide “approach to steady-state” kinetics, this post-transcriptional mechanism is widespread in the temperature-dependent control of gene expression.
September 2016: Temperature regulates splicing efficiency of the cold-inducible RNA-binding protein gene Cirbp. In mammals, body temperature fluctuates diurnally around a mean value of 36°C-37°C. Despite the small differences between minimal and maximal values, body temperature rhythms can drive robust cycles in gene expression in cultured cells and, likely, animals. Here we studied the mechanisms responsible for the temperature-dependent expression of cold-inducible RNA-binding protein (CIRBP). In NIH3T3 fibroblasts exposed to simulated mouse body temperature cycles, Cirbp mRNA oscillates about threefold in abundance, as it does in mouse livers. This daily mRNA accumulation cycle is directly controlled by temperature oscillations and does not depend on the cells’ circadian clocks. Here we show that the temperature-dependent accumulation of Cirbp mRNA is controlled primarily by the regulation of splicing efficiency, defined as the fraction of Cirbp pre-mRNA processed into mature mRNA. As revealed by genome-wide “approach to steady-state” kinetics, this post-transcriptional mechanism is widespread in the temperature-dependent control of gene expression.
• Publisher site • Pubmed • Web of Science • Infoscience
 July 2015: Structure of silent transcription intervals and noise characteristics of mammalian genes. Mammalian transcription occurs stochastically in short bursts interspersed by silent intervals showing a refractory period. However, the underlying processes and consequences on fluctuations in gene products are poorly understood. Here, we use single allele time‐lapse recordings in mouse cells to identify minimal models of promoter cycles, which inform on the number and durations of rate‐limiting steps responsible for refractory periods. The structure of promoter cycles is gene specific and independent of genomic location. Typically, five rate‐limiting steps underlie the silent periods of endogenous promoters, while minimal synthetic promoters exhibit only one. Strikingly, endogenous or synthetic promoters with TATA boxes show simplified two‐state promoter cycles. Since transcriptional bursting constrains intrinsic noise depending on the number of promoter steps, this explains why TATA box genes display increased intrinsic noise genome‐wide in mammals, as revealed by single‐cell RNA‐seq. These findings have implications for basic transcription biology and shed light on interpreting single‐cell RNA‐counting experiments.
July 2015: Structure of silent transcription intervals and noise characteristics of mammalian genes. Mammalian transcription occurs stochastically in short bursts interspersed by silent intervals showing a refractory period. However, the underlying processes and consequences on fluctuations in gene products are poorly understood. Here, we use single allele time‐lapse recordings in mouse cells to identify minimal models of promoter cycles, which inform on the number and durations of rate‐limiting steps responsible for refractory periods. The structure of promoter cycles is gene specific and independent of genomic location. Typically, five rate‐limiting steps underlie the silent periods of endogenous promoters, while minimal synthetic promoters exhibit only one. Strikingly, endogenous or synthetic promoters with TATA boxes show simplified two‐state promoter cycles. Since transcriptional bursting constrains intrinsic noise depending on the number of promoter steps, this explains why TATA box genes display increased intrinsic noise genome‐wide in mammals, as revealed by single‐cell RNA‐seq. These findings have implications for basic transcription biology and shed light on interpreting single‐cell RNA‐counting experiments.
• Publisher site • Pubmed • Web of Science • Infoscience
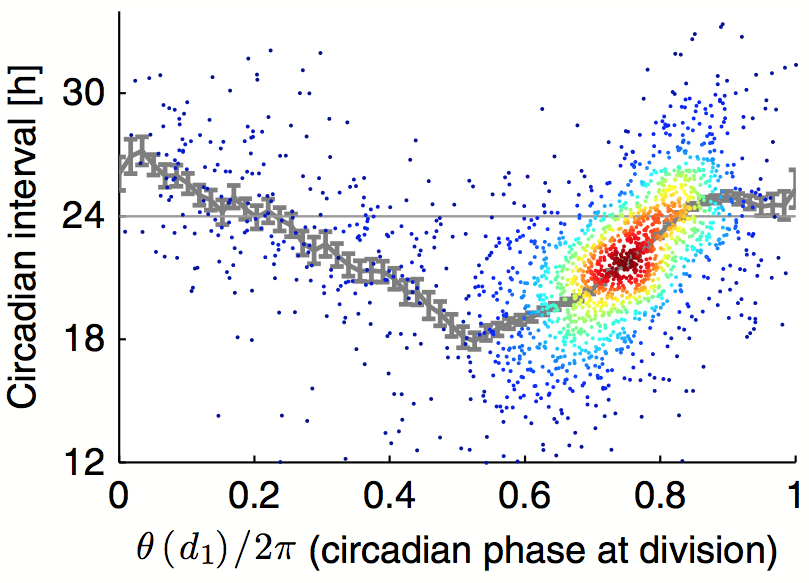 July 2014: Robust synchronization of coupled circadian and cell cycle oscillators in single mammalian cells. Circadian cycles and cell cycles are two fundamental periodic processes with a period in the range of 1 day. Consequently, coupling between such cycles can lead to synchronization. Here, we estimated the mutual interactions between the two oscillators by time‐lapse imaging of single mammalian NIH3T3 fibroblasts during several days. The analysis of thousands of circadian cycles in dividing cells clearly indicated that both oscillators tick in a 1:1 mode‐locked state, with cell divisions occurring tightly 5 h before the peak in circadian Rev‐Erbα‐YFP reporter expression. In principle, such synchrony may be caused by either unidirectional or bidirectional coupling. While gating of cell division by the circadian cycle has been most studied, our data combined with stochastic modeling unambiguously show that the reverse coupling is predominant in NIH3T3 cells. Moreover, temperature, genetic, and pharmacological perturbations showed that the two interacting cellular oscillators adopt a synchronized state that is highly robust over a wide range of parameters. These findings have implications for circadian function in proliferative tissues, including epidermis, immune cells, and cancer.
July 2014: Robust synchronization of coupled circadian and cell cycle oscillators in single mammalian cells. Circadian cycles and cell cycles are two fundamental periodic processes with a period in the range of 1 day. Consequently, coupling between such cycles can lead to synchronization. Here, we estimated the mutual interactions between the two oscillators by time‐lapse imaging of single mammalian NIH3T3 fibroblasts during several days. The analysis of thousands of circadian cycles in dividing cells clearly indicated that both oscillators tick in a 1:1 mode‐locked state, with cell divisions occurring tightly 5 h before the peak in circadian Rev‐Erbα‐YFP reporter expression. In principle, such synchrony may be caused by either unidirectional or bidirectional coupling. While gating of cell division by the circadian cycle has been most studied, our data combined with stochastic modeling unambiguously show that the reverse coupling is predominant in NIH3T3 cells. Moreover, temperature, genetic, and pharmacological perturbations showed that the two interacting cellular oscillators adopt a synchronized state that is highly robust over a wide range of parameters. These findings have implications for circadian function in proliferative tissues, including epidermis, immune cells, and cancer.
• Publisher site • Pubmed • Web of Science • Infoscience
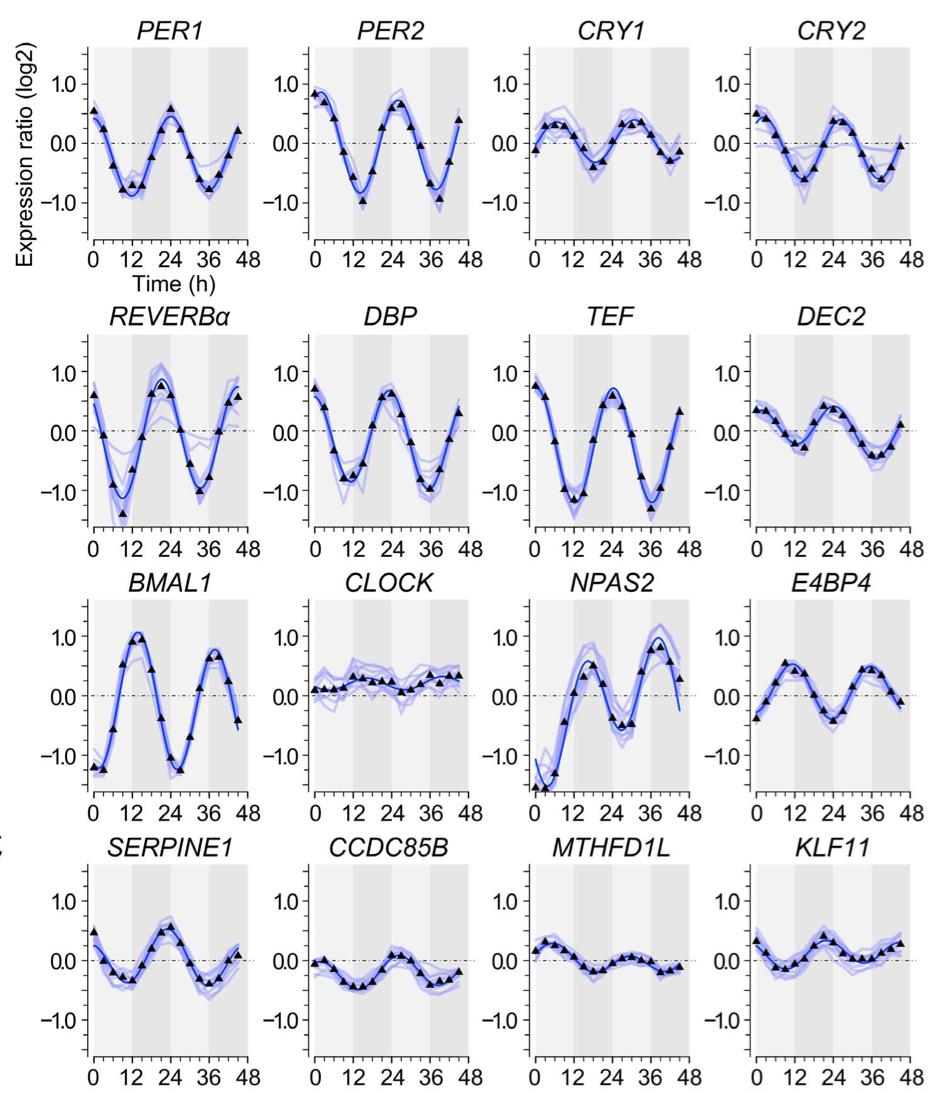 July 2014: Non-Circadian Expression Masking Clock-Driven Weak Transcription Rhythms in U2OS Cells. U2OS cells harbor a circadian clock but express only a few rhythmic genes in constant conditions. We identified 3040 binding sites of the circadian regulators BMAL1, CLOCK and CRY1 in the U2OS genome. Most binding sites even in promoters do not correlate with detectable rhythmic transcript levels. Luciferase fusions reveal that the circadian clock supports robust but low amplitude transcription rhythms of representative promoters. However, rhythmic transcription of these potentially clock-controlled genes is masked by non-circadian transcription that overwrites the weaker contribution of the clock in constant conditions. Our data suggest that U2OS cells harbor an intrinsically rather weak circadian oscillator. The oscillator has the potential to regulate a large number of genes. The contribution of circadian versus non-circadian transcription is dependent on the metabolic state of the cell and may determine the apparent complexity of the circadian transcriptome.
July 2014: Non-Circadian Expression Masking Clock-Driven Weak Transcription Rhythms in U2OS Cells. U2OS cells harbor a circadian clock but express only a few rhythmic genes in constant conditions. We identified 3040 binding sites of the circadian regulators BMAL1, CLOCK and CRY1 in the U2OS genome. Most binding sites even in promoters do not correlate with detectable rhythmic transcript levels. Luciferase fusions reveal that the circadian clock supports robust but low amplitude transcription rhythms of representative promoters. However, rhythmic transcription of these potentially clock-controlled genes is masked by non-circadian transcription that overwrites the weaker contribution of the clock in constant conditions. Our data suggest that U2OS cells harbor an intrinsically rather weak circadian oscillator. The oscillator has the potential to regulate a large number of genes. The contribution of circadian versus non-circadian transcription is dependent on the metabolic state of the cell and may determine the apparent complexity of the circadian transcriptome.
• Publisher site • Pubmed • Web of Science • Infoscience
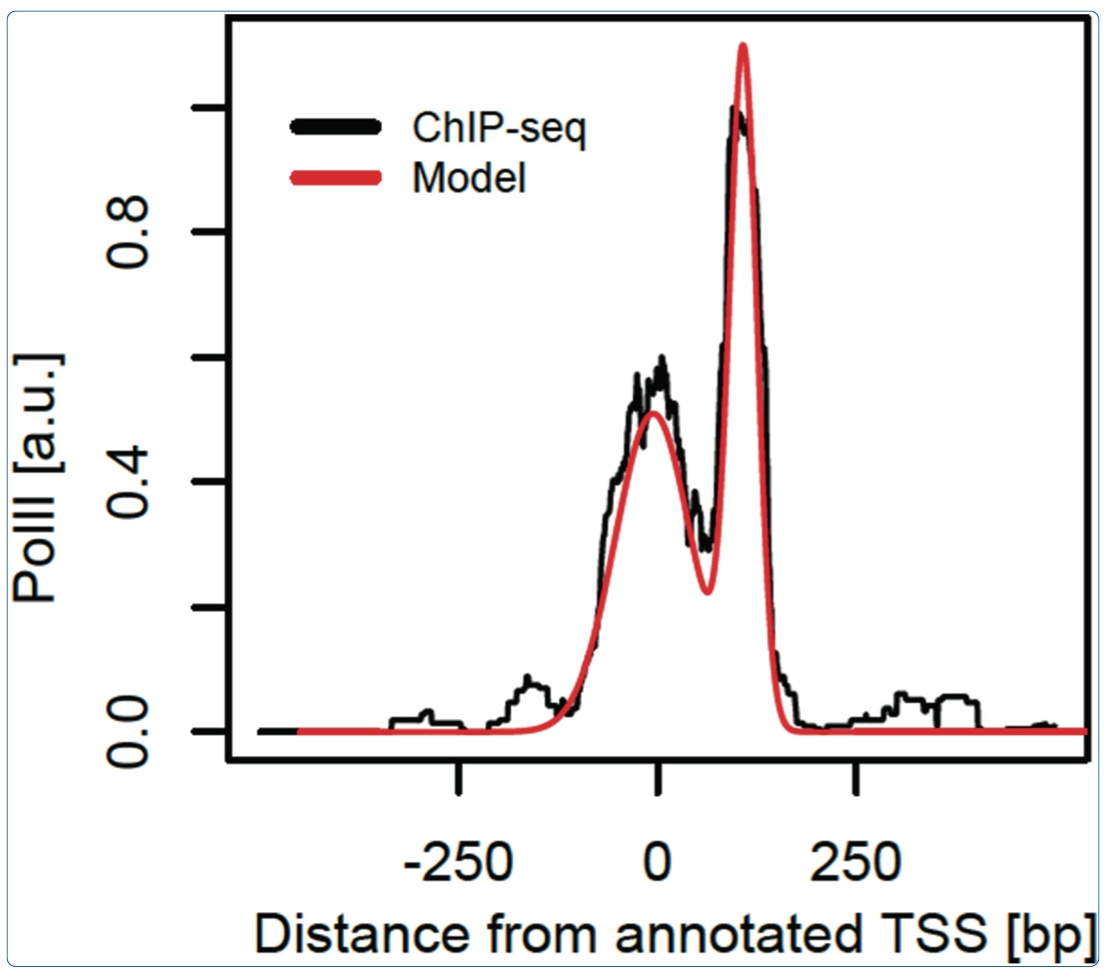 June 2014: Characteristic bimodal profiles of RNA polymerase II at thousands of active mammalian promoters. Background: In mammals, ChIP-seq studies of RNA polymerase II (PolII) occupancy have been performed to reveal how recruitment, initiation and pausing of PolII may control transcription rates, but the focus is rarely on obtaining finely resolved profiles that can portray the progression of PolII through sequential promoter states. Results: Here, we analyze PolII binding profiles from high-coverage ChIP-seq on promoters of actively transcribed genes in mouse and humans. We show that the enrichment of PolII near transcription start sites exhibits a stereotypical bimodal structure, with one peak near active transcription start sites and a second peak 110 base pairs downstream from the first. Using an empirical model that reliably quantifies the spatial PolII signal, gene by gene, we show that the first PolII peak allows for refined positioning of transcription start sites, which is corroborated by mRNA sequencing. This bimodal signature is found both in mouse and humans. Analysis of the pausing-related factors NELF and DSIF suggests that the downstream peak reflects widespread pausing at the +1 nucleosome barrier. Several features of the bimodal pattern are correlated with sequence features such as CpG content and TATA boxes, as well as the histone mark H3K4me3. Conclusions: We thus show how high coverage DNA sequencing experiments can reveal as-yet unnoticed bimodal spatial features of PolII accumulation that are frequent at individual mammalian genes and reminiscent of transcription initiation and pausing. The initiation-pausing hypothesis is corroborated by evidence from run-on sequencing and immunoprecipitation in other cell types and species.
June 2014: Characteristic bimodal profiles of RNA polymerase II at thousands of active mammalian promoters. Background: In mammals, ChIP-seq studies of RNA polymerase II (PolII) occupancy have been performed to reveal how recruitment, initiation and pausing of PolII may control transcription rates, but the focus is rarely on obtaining finely resolved profiles that can portray the progression of PolII through sequential promoter states. Results: Here, we analyze PolII binding profiles from high-coverage ChIP-seq on promoters of actively transcribed genes in mouse and humans. We show that the enrichment of PolII near transcription start sites exhibits a stereotypical bimodal structure, with one peak near active transcription start sites and a second peak 110 base pairs downstream from the first. Using an empirical model that reliably quantifies the spatial PolII signal, gene by gene, we show that the first PolII peak allows for refined positioning of transcription start sites, which is corroborated by mRNA sequencing. This bimodal signature is found both in mouse and humans. Analysis of the pausing-related factors NELF and DSIF suggests that the downstream peak reflects widespread pausing at the +1 nucleosome barrier. Several features of the bimodal pattern are correlated with sequence features such as CpG content and TATA boxes, as well as the histone mark H3K4me3. Conclusions: We thus show how high coverage DNA sequencing experiments can reveal as-yet unnoticed bimodal spatial features of PolII accumulation that are frequent at individual mammalian genes and reminiscent of transcription initiation and pausing. The initiation-pausing hypothesis is corroborated by evidence from run-on sequencing and immunoprecipitation in other cell types and species.
• Publisher site • Pubmed • Infoscience
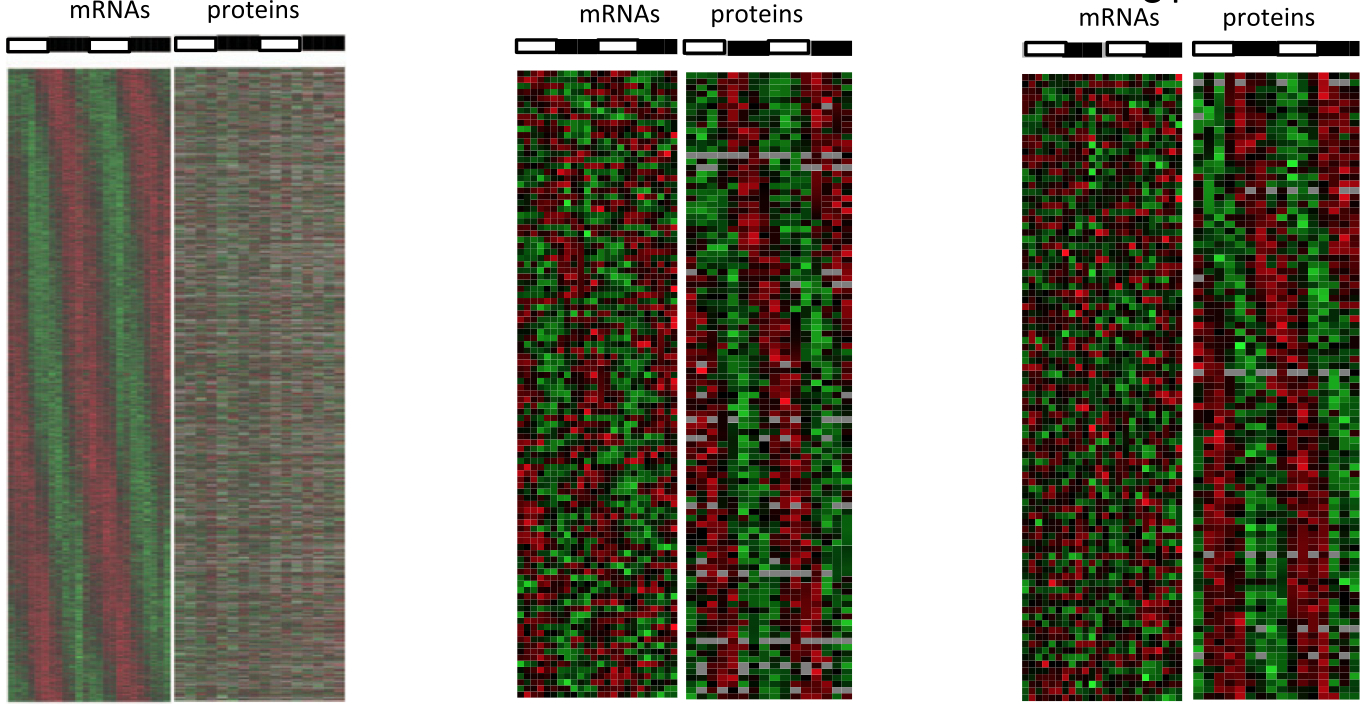 January 2014: Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. Diurnal oscillations of gene expression controlled by the circadian clock underlie rhythmic physiology across most living organisms. Although such rhythms have been extensively studied at the level of transcription and mRNA accumulation, little is known about the accumulation patterns of proteins. Here, we quantified temporal profiles in the murine hepatic proteome under physiological light-dark conditions using stable isotope labeling by amino acids quantitative MS. Our analysis identified over 5,000 proteins, of which several hundred showed robust diurnal oscillations with peak phases enriched in the morning and during the night and related to core hepatic physiological functions. Combined mathematical modeling of temporal protein and mRNA profiles indicated that proteins accumulate with reduced amplitudes and significant delays, consistent with protein half-life data. Moreover, a group comprising about one-half of the rhythmic proteins showed no corresponding rhythmic mRNAs, indicating significant translational or posttranslational diurnal control. Such rhythms were highly enriched in secreted proteins accumulating tightly during the night. Also, these rhythms persisted in clock-deficient animals subjected to rhythmic feeding, suggesting that food-related entrainment signals influence rhythms in circulating plasma factors.
January 2014: Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. Diurnal oscillations of gene expression controlled by the circadian clock underlie rhythmic physiology across most living organisms. Although such rhythms have been extensively studied at the level of transcription and mRNA accumulation, little is known about the accumulation patterns of proteins. Here, we quantified temporal profiles in the murine hepatic proteome under physiological light-dark conditions using stable isotope labeling by amino acids quantitative MS. Our analysis identified over 5,000 proteins, of which several hundred showed robust diurnal oscillations with peak phases enriched in the morning and during the night and related to core hepatic physiological functions. Combined mathematical modeling of temporal protein and mRNA profiles indicated that proteins accumulate with reduced amplitudes and significant delays, consistent with protein half-life data. Moreover, a group comprising about one-half of the rhythmic proteins showed no corresponding rhythmic mRNAs, indicating significant translational or posttranslational diurnal control. Such rhythms were highly enriched in secreted proteins accumulating tightly during the night. Also, these rhythms persisted in clock-deficient animals subjected to rhythmic feeding, suggesting that food-related entrainment signals influence rhythms in circulating plasma factors.
• Publisher site • Pubmed • Web of Science • Infoscience
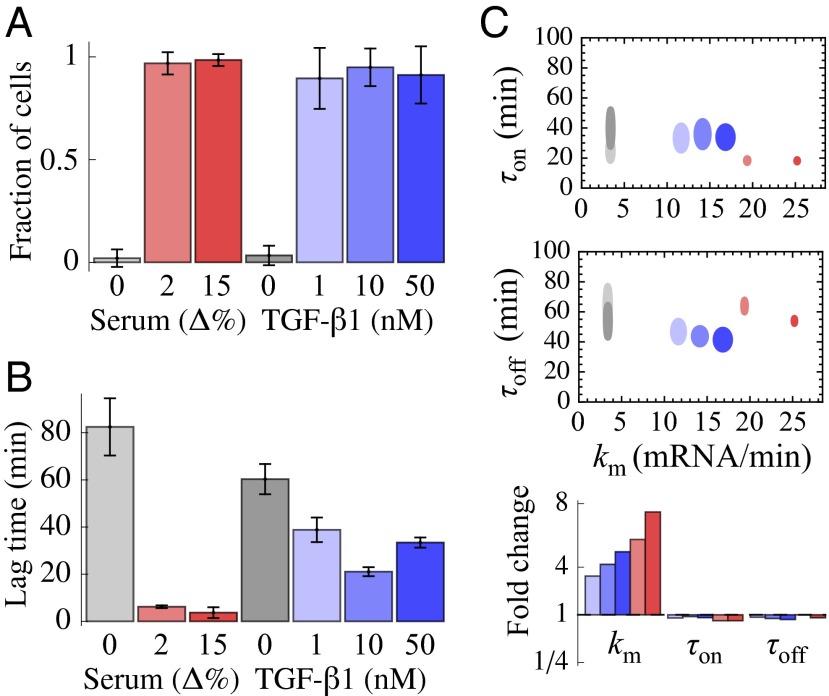
December 2013 : Stimulus-induced modulation of transcriptional bursting in a single mammalian gene. Mammalian genes are often transcribed discontinuously as short bursts of RNA synthesis followed by longer silent periods. However, how these “on” and “off” transitions, together with the burst sizes, are modulated in single cells to increase gene expression upon stimulation is poorly characterized. By combining single-cell time-lapse luminescence imaging with stochastic modeling of the time traces, we quantified the transcriptional responses of the endogenous connective tissue growth factor gene to different physiological stimuli: serum and TGF-beta 1. Both stimuli caused a rapid and acute increase in burst sizes. Whereas TGF-beta 1 showed prolonged transcriptional activation mediated by an increase of transcription rate, serum stimulation resulted in a large and temporally tight first transcriptional burst, followed by a refractory period in the range of hours. Our study thus reveals how different physiological stimuli can trigger kinetically distinct transcriptional responses of the same gene.
• Publisher site • Pubmed • Web of Science • Infoscience
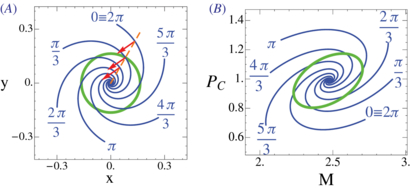
September 2013 : Analysis of precision in chemical oscillators: implications for circadian clocks. Biochemical reaction networks often exhibit spontaneous self-sustained oscillations. An example is the circadian oscillator that lies at the heart of daily rhythms in behavior and physiology in most organisms including humans. While the period of these oscillators evolved so that it resonates with the 24 h daily environmental cycles, the precision of the oscillator (quantified via the Q factor) is another relevant property of these cell-autonomous oscillators. Since this quantity can be measured in individual cells, it is of interest to better understand how this property behaves across mathematical models of these oscillators. Current theoretical schemes for computing the Q factors show limitations for both high-dimensional models and in the vicinity of Hopf bifurcations. Here, we derive low-noise approximations that lead to numerically stable schemes also in high-dimensional models. In addition, we generalize normal form reductions that are appropriate near Hopf bifurcations. Applying our approximations to two models of circadian clocks, we show that while the low-noise regime is faithfully recapitulated, increasing the level of noise leads to species-dependent precision. We emphasize that subcomponents of the oscillator gradually decouple from the core oscillator as noise increases, which allows us to identify the subnetworks responsible for robust rhythms.
• Publisher site • Pubmed • Web of Science • Infoscience
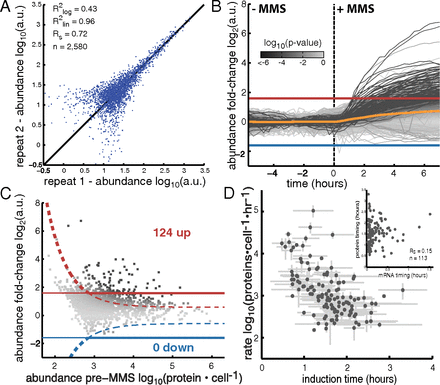 August 2013: A chemostat array enables the spatio-temporal analysis of the yeast proteome. Observing cellular responses to perturbations is central to generating and testing hypotheses in biology. We developed a massively parallel microchemostat array capable of growing and observing 1,152 yeast-GFP strains on the single-cell level with 20 min time resolution. We measured protein abundance and localization changes in 4,085 GFP-tagged strains in response to methyl methanesulfonate and analyzed 576 GFP strains in five additional conditions for a total of more than 10,000 unique experiments, providing a systematic view of the yeast proteome in flux. We observed that processing bodies formed rapidly and synchronously in response to UV irradiation, and in conjunction with 506 deletion-GFP strains, identified four gene disruptions leading to abnormal ribonucleotide-diphosphate reductase (Rnr4) localization. Our microchemostat platform enables the large-scale interrogation of proteomes in flux and permits the concurrent observation of protein abundance, localization, cell size, and growth parameters on the single-cell level for thousands of microbial cultures in one experiment.
August 2013: A chemostat array enables the spatio-temporal analysis of the yeast proteome. Observing cellular responses to perturbations is central to generating and testing hypotheses in biology. We developed a massively parallel microchemostat array capable of growing and observing 1,152 yeast-GFP strains on the single-cell level with 20 min time resolution. We measured protein abundance and localization changes in 4,085 GFP-tagged strains in response to methyl methanesulfonate and analyzed 576 GFP strains in five additional conditions for a total of more than 10,000 unique experiments, providing a systematic view of the yeast proteome in flux. We observed that processing bodies formed rapidly and synchronously in response to UV irradiation, and in conjunction with 506 deletion-GFP strains, identified four gene disruptions leading to abnormal ribonucleotide-diphosphate reductase (Rnr4) localization. Our microchemostat platform enables the large-scale interrogation of proteomes in flux and permits the concurrent observation of protein abundance, localization, cell size, and growth parameters on the single-cell level for thousands of microbial cultures in one experiment.
• Publisher site • Pubmed • Web of Science • Infoscience
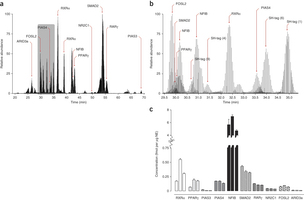 April 2013: Absolute quantification of transcription factors during cellular differentiation using multiplexed targeted proteomics. The cellular abundance of transcription factors (TFs) is an important determinant of their regulatory activities. Deriving TF copy numbers is therefore crucial to understanding how these proteins control gene expression. We describe a sensitive selected reaction monitoring-based mass spectrometry assay that allowed us to determine the copy numbers of up to ten proteins simultaneously. We applied this approach to profile the absolute levels of key TFs, including PPARγ and RXRα, during terminal differentiation of mouse 3T3-L1 pre-adipocytes. Our analyses revealed that individual TF abundance differs dramatically (from ∼250 to >300,000 copies per nucleus) and that their dynamic range during differentiation can vary up to fivefold. We also formulated a DNA binding model for PPARγ based on TF copy number, binding energetics and local chromatin state. This model explains the increase in PPARγ binding sites during the final differentiation stage that occurs despite a concurrent saturation in PPARγ copy number.
April 2013: Absolute quantification of transcription factors during cellular differentiation using multiplexed targeted proteomics. The cellular abundance of transcription factors (TFs) is an important determinant of their regulatory activities. Deriving TF copy numbers is therefore crucial to understanding how these proteins control gene expression. We describe a sensitive selected reaction monitoring-based mass spectrometry assay that allowed us to determine the copy numbers of up to ten proteins simultaneously. We applied this approach to profile the absolute levels of key TFs, including PPARγ and RXRα, during terminal differentiation of mouse 3T3-L1 pre-adipocytes. Our analyses revealed that individual TF abundance differs dramatically (from ∼250 to >300,000 copies per nucleus) and that their dynamic range during differentiation can vary up to fivefold. We also formulated a DNA binding model for PPARγ based on TF copy number, binding energetics and local chromatin state. This model explains the increase in PPARγ binding sites during the final differentiation stage that occurs despite a concurrent saturation in PPARγ copy number.
• Publisher site • Pubmed • Web of Science • Infoscience
 November 2012: Genome-Wide RNA Polymerase II Profiles and RNA Accumulation Reveal Kinetics of Transcription and Associated Epigenetic Changes During Diurnal Cycles. Interactions of cell-autonomous circadian oscillators with diurnal cycles govern the temporal compartmentalization of cell physiology in mammals. To understand the transcriptional and epigenetic basis of diurnal rhythms in mouse liver genome-wide, we generated temporal DNA occupancy profiles by RNA polymerase II (Pol II) as well as profiles of the histone modifications H3K4me3 and H3K36me3. We used these data to quantify the relationships of phases and amplitudes between different marks. We found that rhythmic Pol II recruitment at promoters rather than rhythmic transition from paused to productive elongation underlies diurnal gene transcription, a conclusion further supported by modeling. Moreover, Pol II occupancy preceded mRNA accumulation by 3 hours, consistent with mRNA half-lives. Both methylation marks showed that the epigenetic landscape is highly dynamic and globally remodeled during the 24-hour cycle. While promoters of transcribed genes had tri-methylated H3K4 even at their trough activity times, tri-methylation levels reached their peak, on average, 1 hour after Pol II. Meanwhile, rhythms in tri-methylation of H3K36 lagged transcription by 3 hours. Finally, modeling profiles of Pol II occupancy and mRNA accumulation identified three classes of genes: one showing rhythmicity both in transcriptional and mRNA accumulation, a second class with rhythmic transcription but flat mRNA levels, and a third with constant transcription but rhythmic mRNAs. The latter class emphasizes widespread temporally gated posttranscriptional regulation in the mouse liver.
November 2012: Genome-Wide RNA Polymerase II Profiles and RNA Accumulation Reveal Kinetics of Transcription and Associated Epigenetic Changes During Diurnal Cycles. Interactions of cell-autonomous circadian oscillators with diurnal cycles govern the temporal compartmentalization of cell physiology in mammals. To understand the transcriptional and epigenetic basis of diurnal rhythms in mouse liver genome-wide, we generated temporal DNA occupancy profiles by RNA polymerase II (Pol II) as well as profiles of the histone modifications H3K4me3 and H3K36me3. We used these data to quantify the relationships of phases and amplitudes between different marks. We found that rhythmic Pol II recruitment at promoters rather than rhythmic transition from paused to productive elongation underlies diurnal gene transcription, a conclusion further supported by modeling. Moreover, Pol II occupancy preceded mRNA accumulation by 3 hours, consistent with mRNA half-lives. Both methylation marks showed that the epigenetic landscape is highly dynamic and globally remodeled during the 24-hour cycle. While promoters of transcribed genes had tri-methylated H3K4 even at their trough activity times, tri-methylation levels reached their peak, on average, 1 hour after Pol II. Meanwhile, rhythms in tri-methylation of H3K36 lagged transcription by 3 hours. Finally, modeling profiles of Pol II occupancy and mRNA accumulation identified three classes of genes: one showing rhythmicity both in transcriptional and mRNA accumulation, a second class with rhythmic transcription but flat mRNA levels, and a third with constant transcription but rhythmic mRNAs. The latter class emphasizes widespread temporally gated posttranscriptional regulation in the mouse liver.
• Publisher site • Pubmed • Web of Science • Infoscience
 August 2012: Cold-Inducible RNA-Binding Protein Modulates Circadian Gene Expression Posttranscriptionally. In mammalian tissues, circadian gene expression can be driven by local oscillators or systemic signals controlled by the master pacemaker in the suprachiasmatic nucleus. We show that simulated body temperature cycles, but not peripheral oscillators, controlled the rhythmic expression of cold-inducible RNA-binding protein (CIRP) in cultured fibroblasts. In turn, loss-of-function experiments indicated that CIRP was required for high-amplitude circadian gene expression. The transcriptome-wide identification of CIRP-bound RNAs by a biotin-streptavidin-based cross-linking and immunoprecipitation (CLIP) procedure revealed several transcripts encoding circadian oscillator proteins, including CLOCK. Moreover, CLOCK accumulation was strongly reduced in CIRP-depleted fibroblasts. Because ectopic expression of CLOCK improved circadian gene expression in these cells, we surmise that CIRP confers robustness to circadian oscillators through regulation of CLOCK expression.
August 2012: Cold-Inducible RNA-Binding Protein Modulates Circadian Gene Expression Posttranscriptionally. In mammalian tissues, circadian gene expression can be driven by local oscillators or systemic signals controlled by the master pacemaker in the suprachiasmatic nucleus. We show that simulated body temperature cycles, but not peripheral oscillators, controlled the rhythmic expression of cold-inducible RNA-binding protein (CIRP) in cultured fibroblasts. In turn, loss-of-function experiments indicated that CIRP was required for high-amplitude circadian gene expression. The transcriptome-wide identification of CIRP-bound RNAs by a biotin-streptavidin-based cross-linking and immunoprecipitation (CLIP) procedure revealed several transcripts encoding circadian oscillator proteins, including CLOCK. Moreover, CLOCK accumulation was strongly reduced in CIRP-depleted fibroblasts. Because ectopic expression of CLOCK improved circadian gene expression in these cells, we surmise that CIRP confers robustness to circadian oscillators through regulation of CLOCK expression.
• Publisher site • Pubmed • Web of Science • Infoscience
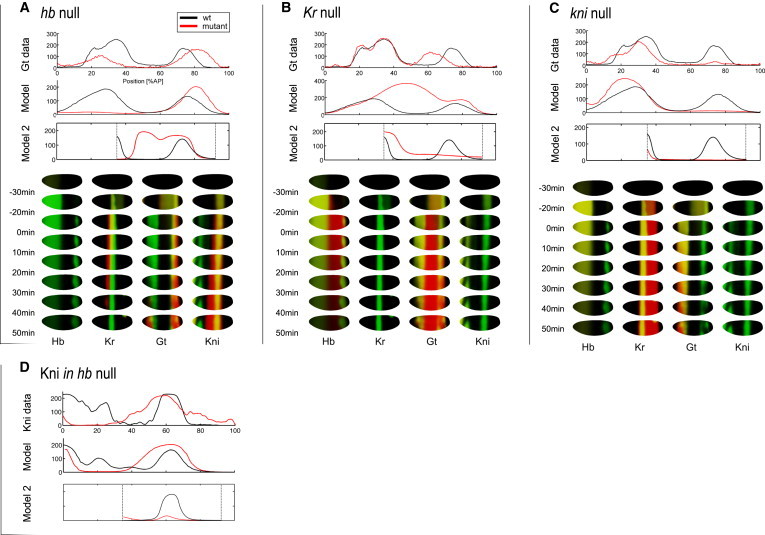 July 2011: Whole-embryo modeling of early segmentation in Drosophila identifies robust and fragile expression domains. Segmentation of the Drosophila melanogaster embryo results from the dynamic establishment of spatial mRNA and protein patterns. Here, we exploit recent temporal mRNA and protein expression measurements on the full surface of the blastoderm to calibrate a dynamical model of the gap gene network on the entire embryo cortex. We model the early mRNA and protein dynamics of the gap genes hunchback, Kruppel, giant, and knirps, taking as regulatory inputs the maternal Bicoid and Caudal gradients, plus the zygotic Tailless and Huckebein proteins. The model captures the expression patterns faithfully, and its predictions are assessed from gap gene mutants. The inferred network shows an architecture based on reciprocal repression between gap genes that can stably pattern the embryo on a realistic geometry but requires complex regulations such as those involving the Hunchback monomer and dimers. Sensitivity analysis identifies the posterior domain of giant as among the most fragile features of an otherwise robust network, and hints at redundant regulations by Bicoid and Hunchback, possibly reflecting recent evolutionary changes in the gap-gene network in insects.
July 2011: Whole-embryo modeling of early segmentation in Drosophila identifies robust and fragile expression domains. Segmentation of the Drosophila melanogaster embryo results from the dynamic establishment of spatial mRNA and protein patterns. Here, we exploit recent temporal mRNA and protein expression measurements on the full surface of the blastoderm to calibrate a dynamical model of the gap gene network on the entire embryo cortex. We model the early mRNA and protein dynamics of the gap genes hunchback, Kruppel, giant, and knirps, taking as regulatory inputs the maternal Bicoid and Caudal gradients, plus the zygotic Tailless and Huckebein proteins. The model captures the expression patterns faithfully, and its predictions are assessed from gap gene mutants. The inferred network shows an architecture based on reciprocal repression between gap genes that can stably pattern the embryo on a realistic geometry but requires complex regulations such as those involving the Hunchback monomer and dimers. Sensitivity analysis identifies the posterior domain of giant as among the most fragile features of an otherwise robust network, and hints at redundant regulations by Bicoid and Hunchback, possibly reflecting recent evolutionary changes in the gap-gene network in insects.
• Publisher site • Pubmed • Web of Science • Infoscience
March 2011: Mammalian Genes Are Transcribed with Widely Different Bursting Kinetics. In prokaryotes and eukaryotes, most genes appear to be transcribed during short periods called transcriptional bursts, interspersed by silent intervals. We describe how such bursts generate gene-specific temporal patterns of messenger RNA (mRNA) synthesis in mammalian cells. To monitor transcription at high temporal resolution, we established various gene trap cell lines and transgenic cell lines expressing a short-lived luciferase protein from an unstable mRNA, and recorded bioluminescence in real time in single cells. Mathematical modeling identified gene-specific on- and off-switching rates in transcriptional activity and mean numbers of mRNAs produced during the bursts. Transcriptional kinetics were markedly altered by cis-regulatory DNA elements. Our analysis demonstrated that bursting kinetics are highly gene-specific, reflecting refractory periods during which genes stay inactive for a certain time before switching on again.
• Publisher site • Pubmed • Web of Science • Infoscience
February 2011: Genome-Wide and Phase-Specific DNA-Binding Rhythms of BMAL1 Control Circadian Output Functions in Mouse Liver. The mammalian circadian clock uses interlocked negative feedback loops in which the heterodimeric basic helix-loop-helix transcription factor BMAL1/CLOCK is a master regulator. While there is prominent control of liver functions by the circadian clock, the detailed links between circadian regulators and downstream targets are poorly known. Using chromatin immunoprecipitation combined with deep sequencing we obtained a time-resolved and genome-wide map of BMAL1 binding in mouse liver, which allowed us to identify over 2,000 binding sites, with peak binding narrowly centered around Zeitgeber time 6. Annotation of BMAL1 targets confirms carbohydrate and lipid metabolism as the major output of the circadian clock in mouse liver. Moreover, transcription regulators are largely overrepresented, several of which also exhibit circadian activity. Genes of the core circadian oscillator stand out as strongly bound, often at promoter and distal sites. Genomic sequence analysis of the sites identified E-boxes and tandem E1-E2 consensus elements. Electromobility shift assays showed that E1-E2 sites are bound by a dimer of BMAL1/CLOCK heterodimers with a spacing-dependent cooperative interaction, a finding that was further validated in transactivation assays. BMAL1 target genes showed cyclic mRNA expression profiles with a phase distribution centered at Zeitgeber time 10. Importantly, sites with E1-E2 elements showed tighter phases both in binding and mRNA accumulation. Finally, analyzing the temporal profiles of BMAL1 binding, precursor mRNA and mature mRNA levels showed how transcriptional and post-transcriptional regulation contribute differentially to circadian expression phase. Together, our analysis of a dynamic protein-DNA interactome uncovered how genes of the core circadian oscillator crosstalk and drive phase-specific circadian output programs in a complex tissue.
• Publisher site • Pubmed • Web of Science • Infoscience